



Alzheimer’s disease (AD) remains a formidable challenge in the medical community, being the leading cause of dementia worldwide. This neurodegenerative disorder affects millions, with epidemiological studies highlighting its high prevalence across diverse populations. Advanced age has been consistently identified as a major risk factor for the development of AD, with experts recognizing three pathological hallmarks: amyloid-beta (Aβ) protein deposition, hyperphosphorylation of Tau protein, and chronic neuroinflammation. These alterations lead to neuronal dysfunction and progressive neurodegeneration.
A significant body of research has identified a cluster of genes associated with AD, including APP, PSEN1, and PSEN2, which play pivotal roles in the disease’s pathogenesis. Mutations in these genes cause monogenic forms of AD, known as Familial AD (FAD), characterized by early onset and autosomal dominant inheritance patterns. These genetic discoveries have provided crucial insights into the molecular mechanisms underlying AD pathogenesis. However, the majority of AD cases fall under the category of sporadic AD (SAD), which shows later symptom onset and lacks familial inheritance patterns.
Genetic Mosaicism and Somatic Mutations
The inability of inherited mutations alone to fully account for AD pathogenesis underscores the significant contribution of environmental factors to disease development. Recent advances in sequencing technologies have revealed that somatic mutations, which occur in non-reproductive cells during an organism’s growth and aging, are prevalent not only in pathological conditions but also in normal cell populations. These mutations are broadly classified into four categories: single nucleotide variants (SNVs), insertions and deletions (Indels), structural variations (SVs), and mitochondrial DNA (mtDNA) mutations.
In the field of oncology, numerous clinically significant somatic mutations have been identified, particularly in driver genes such as EGFR, which play pivotal roles in tumor initiation and progression. Unlike most somatic cells, neurons in the human central nervous system possess limited regenerative capacity. This unique characteristic means that somatic mutations in neurons cannot arise via extensive postnatally occurring cell divisions. Furthermore, because neurons are post-mitotic, they cannot propagate somatic mutations through DNA replication, resulting in exceptionally low variant allele frequency (VAF). This low VAF makes somatic mutations in neurons highly susceptible to background noise, posing significant challenges for detection.
Understanding Somatic Mutations in Neurodegeneration
It is increasingly recognized that somatic mutations play a crucial role in central nervous system (CNS) pathology, with numerous studies establishing compelling associations between these mutations and various neurodegenerative disorders. For instance, somatic BRAF V600E mutations in erythro-myeloid progenitor lineages have been implicated in neurodegenerative processes. There is a growing belief that the somatic expansion of the mutated CAG repeat sequence in brain cells marks the first step in the molecular pathogenesis of Huntington’s disease.
Despite significant advances, the relationship between somatic mutations and AD, one of the most prevalent neurodegenerative disorders, remains poorly understood. Recent research highlights the multifaceted roles of somatic mutations in CNS degeneration, particularly in AD. By elucidating the physiological and pathological implications of these genetic alterations, researchers aim to provide deeper insight into AD pathogenesis through the lens of somatic mutagenesis and propose new avenues for therapeutic intervention and diagnostic development.
Environmental Influences and Lifestyle Factors
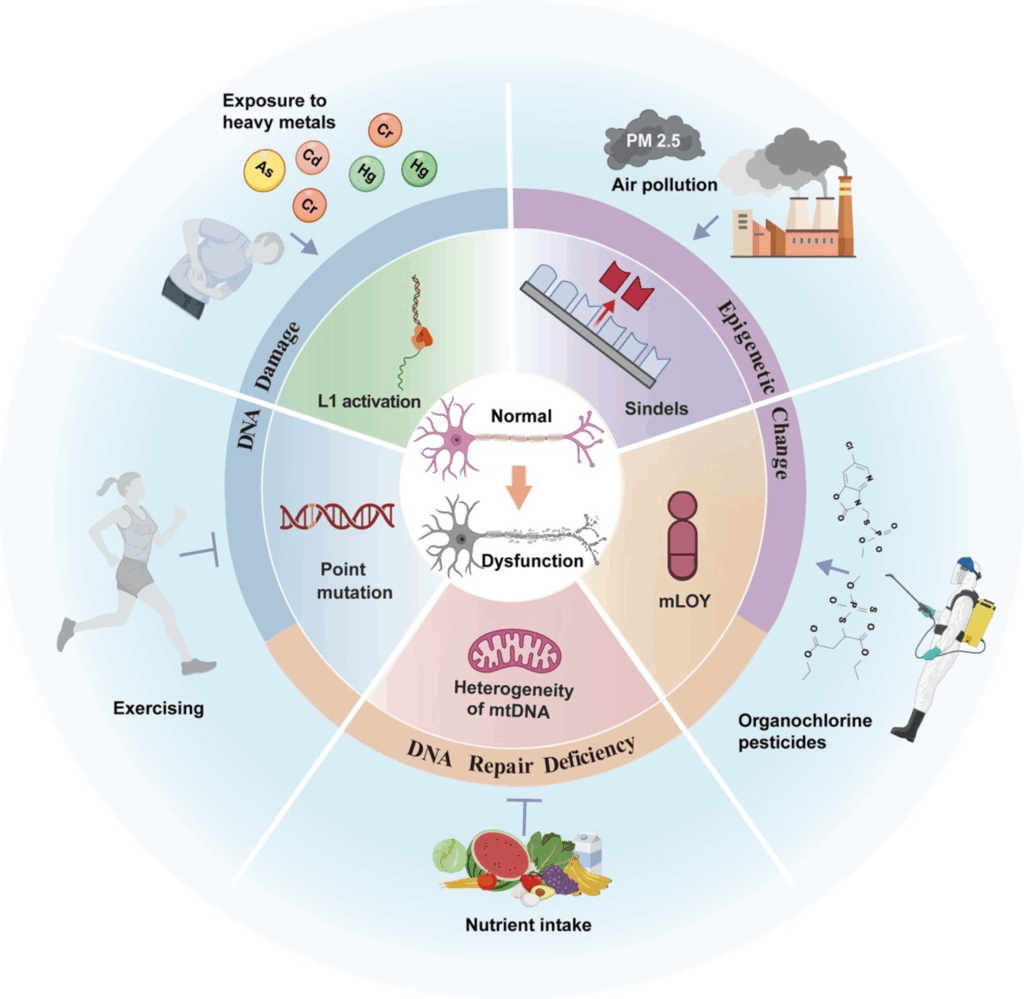
Environmental and lifestyle factors significantly influence the occurrence of somatic mutations. Exposure to heavy metals such as arsenic, cadmium, and mercury increases the risk of AD. Air pollution, especially fine particulate matter (PM2.5), represents a major environmental risk factor for AD. Conversely, regular physical activity reduces the risk of AD and slows cognitive decline in affected individuals. Sufficient intake of vitamin B12 and folate correlates with lower AD risk.
Although several studies have examined how these factors influence AD progression, the underlying mechanisms remain incompletely defined. For example, lead, cadmium, and organophosphate pesticides intensify neuroinflammation, whereas vitamin B12 and folate reduce such inflammation. Physical activity improves mitochondrial structure and function, providing neuroprotective effects. However, current evidence remains fragmented, and a complete mechanistic understanding is still lacking.
Researchers propose that these environmental and lifestyle factors may promote somatic mutations. Metals such as chromium, cadmium, and arsenic induce oxidative stress and DNA repair defects following DNA damage, leading to genomic instability and mutations. Notably, the activity of RNase H2—an enzyme closely associated with Indels formation—is regulated by divalent metal ions such as calcium and magnesium. Even extremely low concentrations of cesium and lithium salts, as well as cellular senescence, have been shown to suppress RNase H2 activity, potentially leading to an increased Indel burden.
Molecular Mechanisms Underlying Somatic Mutations
In the context of AD, DNA damage and repair mechanisms play critical roles in the accumulation of somatic mutations. The robust blood-brain barrier prevents the entry of most genotoxic substances, making endogenous reactive oxygen species (ROS) the primary DNA damage factor in the brain. Oxidation not only directly damages DNA, leading to somatic mutations, but also compromises replication fidelity by impairing DNA polymerase γ.
Neurons are particularly susceptible to DNA damage and frequently sustain double-strand breaks (DSBs) and other lesions. There is substantial evidence supporting an increased incidence of DSBs in patients with mild cognitive impairment (MCI) and AD. Although DSBs are essential for neural development and gene regulation, they can also trigger aberrant repair mechanisms, leading to somatic mutations associated with DNA rearrangements.
Under physiological conditions, a dynamic equilibrium is maintained between DNA damage and repair to preserve genomic stability. Neurons, which lack proliferative capacity, heavily rely on DNA repair to ensure genomic accuracy. Researchers estimate that a single neuron may undergo up to a billion DNA repair events during a person’s lifetime. When the extent of damage exceeds the repair capacity, genetic mutations and apoptosis can occur, phenomena that become increasingly prevalent in senescent cells.
In conclusion, understanding the complex interplay between genetic mosaicism, environmental influences, and molecular mechanisms is crucial for unraveling the pathogenesis of Alzheimer’s disease. As research continues to shed light on these intricate processes, new therapeutic strategies and diagnostic tools may emerge, offering hope for those affected by this devastating condition.


